一、等离子体氧化电解液的优化
对硅酸钠-磷酸钠混合体系电解液进行对比优化实验,以测得氧化时间内起弧电压,终止电压,时间与电压关系等参数以及陶瓷层厚度。多聚磷酸钠Na3P5O10浓度恒定为15 g/l。
经过大量尝试性实验发现,当Na2SiO3浓度从4 g/l变到10 g/l时,有利于陶瓷层的生长,当其浓度较低时,成膜速率较低,并且基本保持不变,约0.6μm/min左右;当Na2SiO3浓度增加时,成膜速率变化较大,升至2.0μm/min,而陶瓷层变得粗糙。因此选定Na2SiO3浓度为6 g/l,8 g/l两种进行实验分析。
定的范围内,随Na2SiO3浓度的增大膜层厚度增加,随着KOH浓度的升高,氧化的起弧电压逐渐降低。根据如上正交实验得出,在硅酸钠体系下,LS2号电解液,即成份为Na3P5O10为15 g/l,Na2SiO3为8 g/l,KOH为2 g/l时,生成的陶瓷层,厚度大于其它编号的陶瓷层且均匀,总体质量和外观均好于其它编号的陶瓷层。
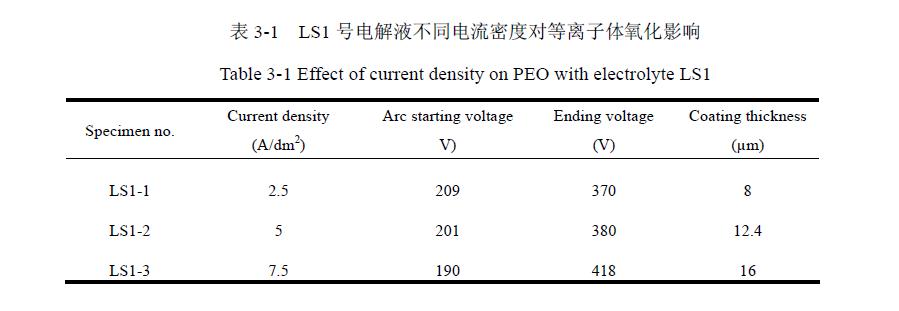
二、陶瓷层厚度与处理时间及电流密度的关系
由图3-5可以看出,较短的时间内,陶瓷层厚度线性增加,并且随着电流密度的增加,陶瓷层的生长速率加大;随时间延长,较大电流密度的氧化陶瓷膜层厚度变化趋于平缓,较小电流密度的膜层依然呈线性趋势,因此在较大的电流密度下,等离子体氧化过程处理效率较高。但当电流密度过大或处理时间过长时,试样表面会产生红色的火花或有爆鸣声,此时对陶瓷层有很大破坏作用,会导致陶瓷层产生过烧,表面发黑,放电孔变大表面质量难以保证。如果电流密度过小,则增加了反应时间,又降低了处理效率,且容易使基体升温过高而影响反应进行。因此电流密度范围为2.5 A/dm2~7.5 A/dm2为宜,最终确定为5 A/dm2;强化时间确定为15 min。
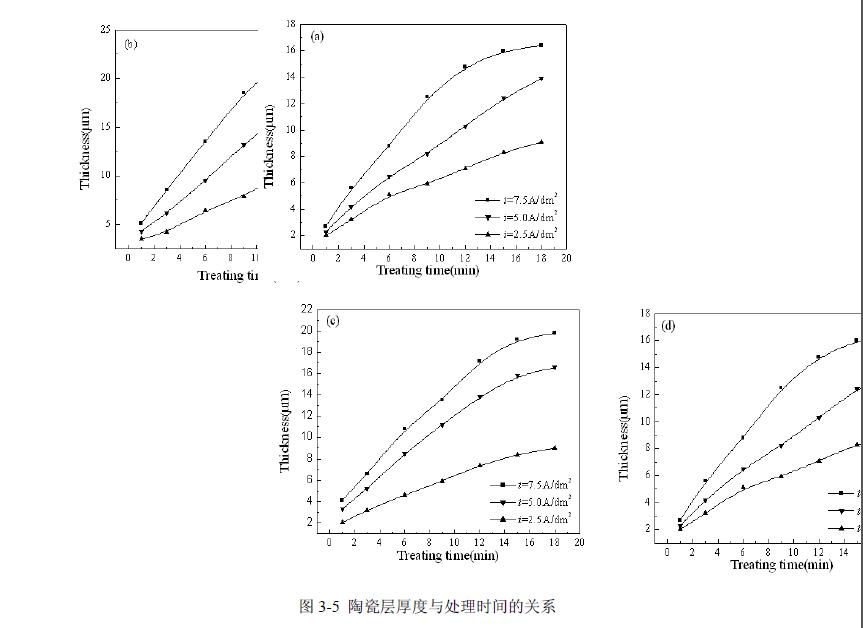
三、电流电压随处理时间变化及能耗
通过图3-6曲线可以看出,在等离子体氧化过程中,当电流恒定时,电压总体随氧化时间的延长而增加,在等离子体氧化初期,电压呈线性上升,且速度较快,随时间延长,电压的上升变得平缓;当电压恒定时,随处理时间的延长,电流逐渐变小,氧化初期电流降落较快,后期变缓。
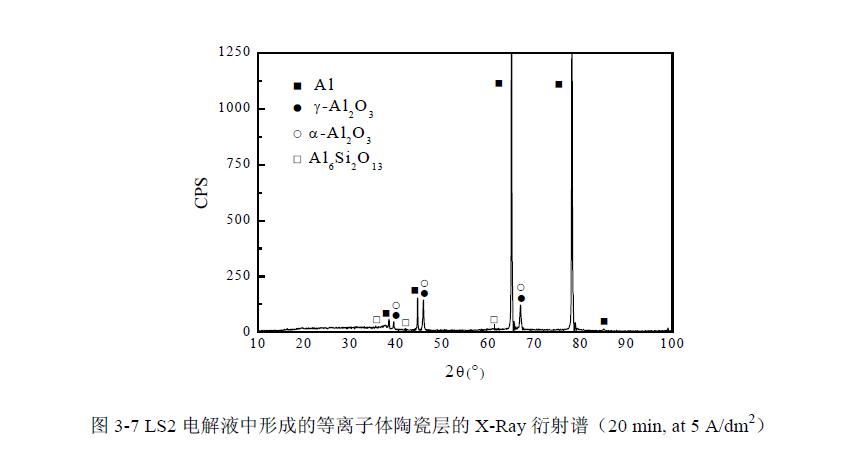
四、陶瓷层的相组成
图3-7为LS2号电解液中,电流密度为5 A/dm2,强化时间为20 min条件下获得陶瓷层的X-射线谱。可明显看出衍射谱中出现了较强的铝基体的衍射峰。此时陶瓷膜层厚度约为20μm左右,而正常X射线衍射的检测深度可达76μm,因此X射线可以直接穿过陶瓷层打在基体铝上。直流条件下包含Na2SiO3的溶液中形成的陶瓷层是由大量的γ-Al2O3和大量的非晶态氧化硅组成,是由于大量的氧化硅在放电通道口沉积,抑制了高温相的α-Al2O3在试样表面形成。而本试验在交流条件下形成的陶瓷层组成研究发现,陶瓷层主要由γ-Al2O3,α-Al2O3和Al6Si2O13相组成。其中膜层内部的α-Al2O3的生成可能由于硅酸根离子只能在放电通道口沉积,不能进入放电通道内部放电,因此放电通道内部温度较高,有利于高温相α-Al2O3的生成。
五、陶瓷层表面与断面形貌分析
图3-8为陶瓷层的截面形貌,可以看出陶瓷层较为致密,具有明显的内外两层结构,膜层与基体之间没有清晰的分界线,结合很好,界面上不存在大的孔洞。A为铝基体,B为内部紧贴基体的致密层,组织致密,与基体之间结合紧密,没有贯穿性的孔洞存在;C为陶瓷层外侧的较为疏松的组织,在最靠近最外侧的地方还存在一些类似粉体的局部区域。
图3-9中试样陶瓷层表面所分布的黑色孔洞即为放电通道。各图中可以清晰的看到形状不规则的放电通道,周围的熔融物冷却堆积成“熔岩”状,整个表面显得不连续。随氧化时间延长,表面变得连续且光滑,放电通道的尺寸变大,数量减少。
当达到45 min时,如图3-9(g)可以明显看到表面的陶瓷层出现裂纹,此时表面质量下降,这也证明了随着等离子体放电的进行,放电通道逐渐被填充,击穿变得困难,后期只能在特定的区域进行放电,此时放电火花体积较大,会导致陶瓷层的剥落,因此陶瓷层厚度有变小的趋势,即陶瓷层不可能无限制的增厚。也可以说,在给定电解液条件下陶瓷层会得到一个最终的厚度。
从上述研究结果可以看出,等离子体电解氧化陶瓷层内存在一定数量的孔隙,对耐蚀性不利,这是该技术存在的一个主要缺点之一。有文献报道,WO42-、PO43-、BO33-、Cr2O72-等可以调节膜层的生长速率,制成性能优异的陶瓷层;F-的加入可以获得强度、硬度适中,结合力、耐蚀性、电绝缘性均优良的陶瓷层;甘油的加入使尖端产生配位物,使沉积更加稳定;在等离子体电解氧化电解液中加入石墨和MoS2等粉末后,等离子体电解氧化陶瓷层形成过程中,能提高陶瓷层的耐磨性能,达到表面改性的目的。本文分别选用磷酸盐和硅酸盐体系电解液为基础,研究不同添加剂和粉末颗粒对等离子体氧化陶瓷层的影响。最后,将超声波引入等离子体电解氧化过程中,研究其对陶瓷层组织性能的强化作用。
http://www.dgzhenghang.net



